.png)
Introduction
After the Second World War, a study called the Framingham Heart Study was initiated. The study is named after the city of Framingham, Massachusetts. Here, a group of researchers recruited 5209 residents of this city between the age of 30 and 62. These people were then regularly evaluated in order to determine risk factors for cardiovascular disease. At the time, very little was known about risk factors for cardiovascular disease. Hypertension? Yeah, they knew it was kind of bad but didn't really have a clue about what would constitute a hypertensive value. For example, at the time, "systolic pressures of up to 210 mmHg and diastolic pressures of 110 mmHg can be regarded as the upper limits of normality" [1]. For those of you who don't know what these numbers mean: this is now deemed a hypertensive crisis and it increases your risk for cardiovascular diseases (and other things) immensely. Such values require direct treatment. Needless to say, they simply didn't have much of a clue at all at the time.
The first results of the Framingham Heart Study were published in 1957 [2]. Herein, they identified three risk factors for cardiovascular disease: hypertension, obesity, and hypercholesterolemia (high cholesterol). Their definition of hypertension is closer to what it is today, with a systolic blood pressure threshold of 160 mmHg and a diastolic threshold of 100 mmHg. More interesting for this article, however, was their identification of hypercholesterolemia as a risk factor for cardiovascular disease. This spawned research into cholesterol, and other lipids transported in the bloodstream, for many decades to come.
Research to date has focused mainly on total cholesterol, low-density lipoprotein (LDL) cholesterol, high-density lipoprotein (HDL) cholesterol, apolipoprotein B (apoB), apolipoprotein A1 (apoA1) and lipoprotein(a) [Lp(a)]. Let's go over each of these and see how anabolic steroids (AAS) influence them.

LDL and HDL cholesterol
LDL and HDL cholesterol likely don't require much of an introduction. They're often portrayed in the media as being 'bad' and 'good' cholesterols, respectively. High LDL cholesterol is associated with an increase in cardiovascular disease; whereas high HDL cholesterol is associated with a decrease in cardiovascular disease. And not only is high LDL cholesterol associated with an increase in cardiovascular disease, its causal link has been firmly established [3]. Treating LDL cholesterol is therefore a crucial part of cardiovascular risk management. Pharmacologically, this is primarily achieved by a class of drugs called statins. These inhibit the rate-limiting enzyme (HMG-CoA reductase) in the biosynthesis of cholesterol. Typically, around two-thirds of the total body input comes from biosynthesis, with the remainder coming from the diet [4]. The result of this inhibition is that LDL cholesterol decreases. Subsequently, the risk for cardiovascular disease drops. Statins will be covered in a future article.
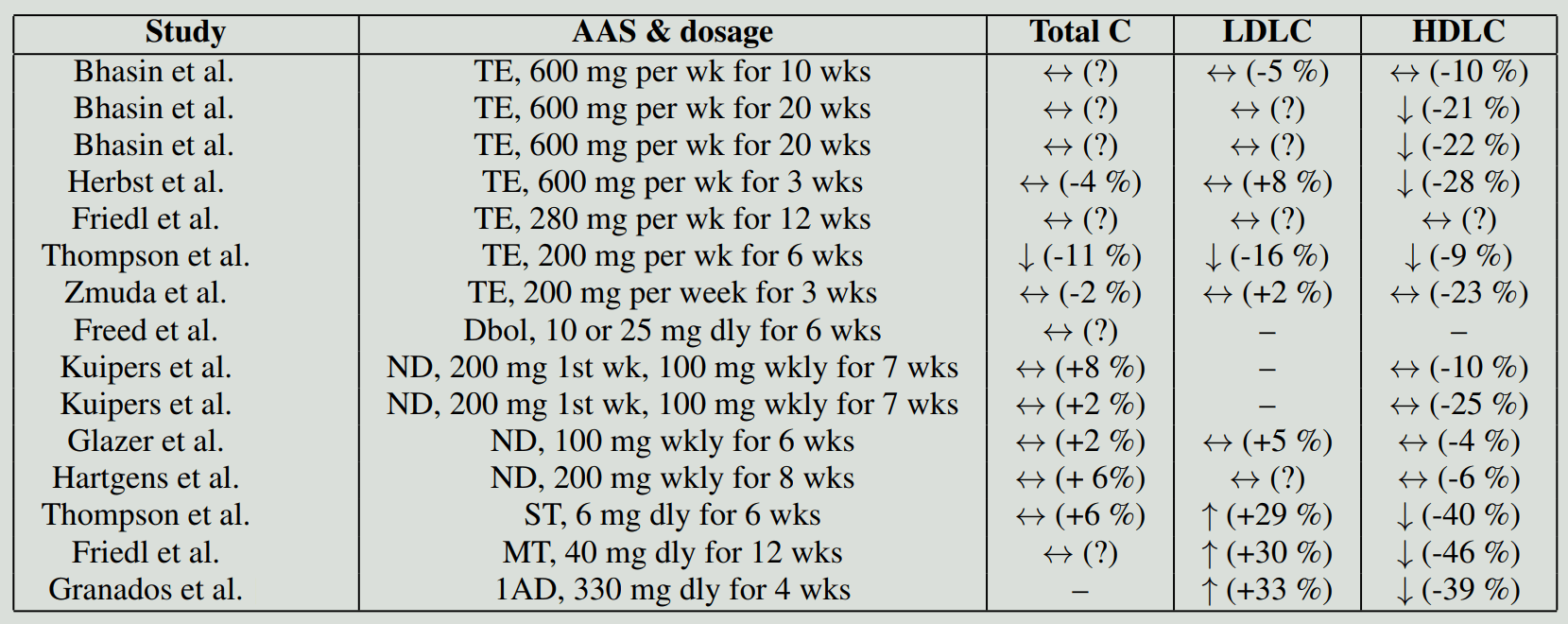
So, how do AAS affect these? Let's start by looking at controlled studies. (I've chosen not to cover trials involving participants with some disease state, including hypogonadism.) If we grab Bhasin's 1996 paper, which I covered extensively in a previous post named How well do anabolic steroids work?, we see that 10 weeks of 600 mg testosterone enanthate weekly had no significant effect on total, LDL or HDL cholesterol [5]. However, it's noteworthy that in Bhasin's 2001 paper, with a similar protocol, a 21 % decrease in HDL cholesterol was observed in the group receiving 600 mg testosterone enanthate weekly for 20 weeks [6]. Total and LDL cholesterol remained unchanged, however. Possible explanations for this might be the longer study duration, but also the fact that participants didn't exercise in the 20-week study. Exercise might increase HDL cholesterol [7] and this effect might have negated any testosterone-induced decrease, but this is speculative. Similar results are seen in older men receiving 600 mg testosterone enanthate weekly for 20 weeks [8]. Notably, an RCT lasting 3 weeks in elderly, obese, eugonadal men demonstrated a significant decrease in HDL cholesterol (-28 %) after only 3 weeks of 600 mg testosterone enanthate weekly [9].
Additionally, in another trial, a small but statistically significant and persistent decrease in HDL cholesterol was observed in men receiving 200 mg testosterone enanthate weekly for 12 months [10]. Unfortunately, that trial lacked a control group. Similarly, another trial (unfortunately also lacking a control group) also found a small decrease in HDL cholesterol after 6 weeks of 200 mg testosterone enanthate weekly [11]. This trial also had a look at HDL cholesterol subfractions HDL2 cholesterol and HDL3 cholesterol. The HDL2 cholesterol particles are larger and of a lower density than the HDL3 cholesterol particles. Without going into detail, there's some evidence suggesting that HDL2 cholesterol is more predictive of cardiovascular disease risk than HDL cholesterol or HDL3 cholesterol [12]. (To be clear: a higher HDL2 cholesterol is better, i.e., an inverse correlation.) The HDL cholesterol decrease caused by testosterone enanthate was in the HDL3 cholesterol subfraction. In contrast, the earlier trial I mentioned with elderly, obese, eugonadal men showed significant decreases compared to baseline in both subfractions with only the decrease in HDL2 cholesterol being significant compared to the placebo [9]. This difference might be related to differences in dosage, study duration or characteristics of the trial participants between the studies.
In another paper, two randomized trials are reported [13]. In each of them, bodybuilders were randomized to receive either a placebo or nandrolone decanoate (200 mg in the first week and then 100 mg weekly for 7 weeks). However, one of these two was carried out in a crossover fashion. It was only in the crossover trial that a significant decrease in HDL cholesterol (-25 %) was found after the nandrolone decanoate injections. While taking the placebo, HDL cholesterol remained practically unchanged (+3 %). The other trial found a nonsignificant 10 % decrease in the nandrolone group, but the placebo group also experienced an 8 % decrease. Total cholesterol remained unchanged in both trials and LDL cholesterol wasn't measured. Additionally, the decrease found in the crossover trial was only significant compared to the baseline; it wasn't significantly different compared to the placebo. This might be because the trial had a low sample size of only 5 participants. There's another trial in which 21 male subjects received 100 mg nandrolone decanoate weekly for 6 weeks, but it lacked a control group [14]. In this trial no significant effect on LDL cholesterol (+5 %) and HDL cholesterol (-4 %) was found compared to the baseline.
Hartgens et al. ran a randomized trial of 8 weeks in which bodybuilders received 200 mg nandrolone decanoate weekly [15]. The authors reported no significant change in total cholesterol, LDL cholesterol, and HDL cholesterol. This trial also evaluated the HDL2 cholesterol and HDL3 cholesterol subfractions. However, there was no significant change in either.
Another trial that measured the HDL2 cholesterol and HDL3 cholesterol subfractions compared the 17α-alkylated oral AAS stanozolol with testosterone [11]. In the study, the participants (weight lifters) were pseudorandomized (they assigned every other subject to one or the other, which isn't really a good form of randomization) to either 6 mg stanozolol daily or 200 mg testosterone enanthate weekly for 6 weeks in a crossover design. The stanozolol group saw their HDL cholesterol decline by 40 % compared to baseline. (The abstract reports a decline of 33 %, but the data in Table 1 simply doesn't support this. I suspect they made a mistake and calculated the decline after 1 week instead of 6.) This decline was most prominent in HDL2 cholesterol, with a staggering decrease of 78 %. (Again, they note a different number in the abstract. I've calculated it based on their numbers in Table 1.) HDL3 cholesterol declined by 22 %. So, the largest decline was seen in HDL2 cholesterol, which is the subfraction that might be more predictive of cardiovascular heart disease risk than HDL cholesterol or HDL3 cholesterol. Or, in other words, it's bad news. Additionally, the stanozolol group saw their LDL cholesterol increase by 29 % compared to baseline.
The effect of testosterone enanthate in this trial on serum lipids was less unfavorable than stanozolol. The testosterone group saw their HDL cholesterol decrease by only 9 % compared to baseline. Notably, there was no significant decrease in HDL2 cholesterol, only in HDL3 cholesterol. Additionally, LDL cholesterol decreased in this group by 16 %. What I feel should be highlighted are the dosages used. Remember, the stanozolol group only received 6 mg daily (i.e., 42 mg weekly) and their lipids went to shit. The testosterone group received 200 mg weekly with more desirable changes in serum lipids.
Lastly, Granados et al. ran a randomized trial with the oral prohormone 1-androsterone [16]. Seventeen resistance-trained men received either a placebo or 330 mg 1-androsterone daily for 4 weeks. Total cholesterol remained unchanged, but LDL cholesterol showed an increase of 33 % and HDL cholesterol showed a decrease of 39 %.
While randomized trials are obviously very nice to have in order to draw some conclusions, they do suffer from some major drawbacks, namely, the dosages and timespan. In the 1996 trial by Bhasin et al., one group received 600 mg testosterone enanthate weekly. While certainly a dosage used by bodybuilders, it's kind of a 'beginners' dosage, and 10 weeks might have been too short to result in changes in serum cholesterol concentrations—especially in light of the other trials, most notably the 2001 and 2005 Bhasin ones, which suggest a decrease in HDL cholesterol. Nevertheless, it certainly seems testosterone enanthate is a relatively less harmful bet in regard to these two blood values: the impact seems relatively small. Because Bhasin's group measured cholesterol in groups receiving a variety of dosages (25, 50, 125, 300, and 600 mg testosterone enanthate weekly), they could evaluate whether there's a dose-response relationship [17]. They found a moderate inverse relationship (r = -0.40) between testosterone levels and HDL cholesterol. Or, in other words, the higher the testosterone dosage, the lower the resulting HDL cholesterol. It's hard to say how this extrapolates beyond 600 mg weekly, but the prospect isn't good.
Interestingly, aromatization of testosterone might also underlie its modest effect on HDL cholesterol compared to other AAS [18, 19]. Friedl et al. randomized a small group of men to one of three treatment groups: 280 mg testosterone enanthate weekly, 280 mg testosterone enanthate weekly + daily testolactone (an old aromatase inhibitor), or 20 mg methyltestosterone twice daily [18]. The trial lasted 12 weeks. As expected, there was a firm decrease in HDL cholesterol in the methyltestosterone group (-56 %). (I don't know why, but a lot of the numbers in the abstract don't match up with the numbers in the results section or the figures.) The group receiving testosterone enanthate without the aromatase inhibitor showed no significant reduction in HDL cholesterol, whereas the group that combined it with an aromatase inhibitor did see a significant reduction (-25 %). This suggests that the aromatization of testosterone, leading to an increase in estrogen, counteracts the negative effect androgens might have on HDL cholesterol.
A later randomized crossover trial also tested the HDL2 and HDL3 cholesterol subfractions [19]. The participating weight lifters received 200 mg testosterone enanthate weekly, testolactone daily, or both together. Testolactone alone had no significant impact on any serum lipid. Testosterone alone, however, did show a significant decrease in HDL cholesterol compared to baseline (-16 %). While it also showed a decrease in the HDL2 cholesterol subfraction (-23 %), this wasn't statistically significant. But it did show a significant decrease in HDL3 cholesterol (-13 %). Notably, in combination with testolactone, a slightly larger decrease in HDL cholesterol was observed (-20 %). HDL2 cholesterol also decreased significantly compared to baseline by 30 % and HDL3 cholesterol by 13 %. It should be noted that, while the decreases were slightly larger when combined with testolactone, no significant differences were found between these two groups. Another drawback of this study is that it only lasted 3 weeks. It might be that its full effect on cholesterol levels takes longer than that.
The decrease in HDL cholesterol is thought to be caused by an increase in the activity of hepatic lipase (HL) [9, 16, 18, 19]. HL is an enzyme produced primarily in the liver. It liberates fatty acids from triacylglycerol and phospholipids that are part of lipoproteins, including HDL cholesterol. As a result, the HDL particles end up smaller. Essentially this is turning the larger HDL2 particles into smaller HDL3 particles. These smaller HDL particles are more susceptible to further breakdown. As a consequence, the overactivity of hepatic lipase leads to a reduction of HDL cholesterol.
But how bad is this decrease in HDL cholesterol in terms of developing cardiovascular disease? This is a hard question to answer, as there's no research linking the AAS-induced decrease in HDL cholesterol to cardiovascular disease. And, while pharmacologically lowering LDL cholesterol decreases the risk for cardiovascular disease, increasingHDL cholesterol in general doesn't seem to do this. Obviously, there is a clear relation between HDL cholesterol and cardiovascular risk, but raising HDL cholesterol with drugs just doesn't seem to cut it [20]. Conversely, it's false to assume that a drug, such as AAS, that decreases HDL cholesterol also increases cardiovascular risk. Because of this disconnect between a drug's effect on HDL cholesterol and cardiovascular risk, research is now more aimed at HDL cholesterol function instead of HDL cholesterol itself. Notably, cholesterol efflux capacity has gained some traction [21]. Cholesterol efflux capacity refers to the capacity of HDL particles to accept cholesterol from cells, in particular, macrophages [22]. Macrophages are cells of the immune system that are intimately involved in the development of atherosclerosis. They are phagocytic cells, meaning that they 'eat' other substances or cells by engulfing them. In regard to atherosclerosis, they engulf (oxidized) LDL cholesterol (an excellent review on this topic is written by Moore & Tabs [23]). These cholesterol-laden cells can accumulate in the arterial wall—a key element of atherosclerosis development. HDL particles can take this cholesterol up again. They can remove it from the macrophages (cholesterol efflux) in the arterial wall and transport it back to the liver. Macrophages can release their cholesterol by simple diffusion or through transporter proteins. The most important transporter that moves cholesterol out of the cholesterol-enriched macrophages is likely the ATP-binding cassette transporter A1 (ABCA1) [24]. Researchers think that cholesterol efflux from macrophages is one of the main ways HDL cholesterol protects arteries from deteriorating.
Some research has looked at the effect of testosterone administration on HDL cholesterol efflux capacity. In a noncontrolled trial, older hypogonadal men were randomized to receive testosterone replacement therapy with or without the 5α-reductase inhibitor dutasteride [25]. After 3 months, testosterone was significantly increased in both groups. However, HDL cholesterol remained unchanged. Similarly, HDL cholesterol efflux remained unchanged.
A recent double-blind RCT also investigated the effect of testosterone (and estradiol) on HDL cholesterol efflux capacity [26]. Healthy men were medically castrated with a GnRH receptor antagonist (acyline) and randomized to one of four groups. One group received a placebo gel, another received a low-dose testosterone gel, and yet another received a full replacement dose testosterone gel. In order to discern the effects of testosterone and estrogen, the fourth group received the full replacement dose testosterone gel in conjunction with an aromatase inhibitor letrozole).
HDL cholesterol remained unchanged in the two groups receiving the full replacement dose of testosterone. HDL cholesterol increased a bit in the placebo group and the group receiving a low dose of testosterone. Recall that they all got their testosterone suppressed by the administration of acyline, hence the change in the placebo group. The HDL capacity to efflux cholesterol from macrophages remained unchanged in all four groups. A small increase was found in ABCA1 efflux capacity in the group also receiving an aromatase inhibitor. Nevertheless, there were no statistical significant differences between the groups. Moreover, the authors also demonstrated a positive (weak; β = 0.33 per 10 pg/mL) correlation between macrophage cholesterol efflux capacity and serum estradiol. I suppose the small sample sizes might have lead to no difference between the groups in that regard.
In contrast with the above results, a cross-sectional study did show an impairment of efflux capacity in AAS users [27]. The researchers compared AAS users with age-matched, strength-trained nonusers and a group of sedentary controls. The AAS users had used AAS for an average of 8 years and were using dosages of—if I'm interpreting their table correctly—roughly 2.5 g weekly, including an average of 350 mg stanozolol weekly. HDL cholesterol in the AAS users was less than half of that in the other two groups. LDL cholesterol was also elevated compared to the other two. The HDL capacity to efflux cholesterol from macrophages was about 13 % lower in the AAS users compared to the strength-trained nonusers. Unfortunately, it's impossible to say to what extent the usage of an oral steroid affected this. In fact, it's impossible to say to what extent this was influenced by AAS usage per se given it was a cross-sectional study. What is also interesting to note, is that one in four of the AAS group showed signs of subclinical coronary artery disease. None of the subjects in the other groups did. And they shouldn't, because it's relatively uncommon given the age of the subjects (they had a mean age of 29 years old, with the oldest seemingly being 43 years of age).
Nevertheless, it therefore seems AAS administration might affect efflux capacity negatively. A decrease implies an increase in cardiovascular disease risk. Problematic is that both randomized trials that evaluated efflux capacity also didn't show a change in HDL cholesterol. The question of whether the decrease in HDL cholesterol induced by AAS under certain circumstances parallels a decrease in efflux capacity therefore remains. The results of the cross-sectional study should be interpreted with great caution due to the nature of the study. Cross-sectional studies are vulnerable to various biases that can influence the results. The correlation should thus be seen for what it is: a correlation.
While it's too early to say that the AAS-induced decrease in HDL cholesterol increases the risk of CVD, it's equally premature to say it doesn't increase the risk of CVD. Nevertheless, no trial is available that has evaluated the effect of a supraphysiological dosage of testosterone (or any other AAS) on HDL cholesterol efflux capacity.
Apolipoprotein A1 and B
While LDL and HDL cholesterol are very well known in the lay public, the apolipoproteins A1 and B are less known. So what are these apolipoproteins? They're proteins associated with lipoproteins, such as LDL and HDL cholesterol (see figure below). More specifically, apolipoprotein A1 is associated with HDL cholesterol, but not with LDL cholesterol. (A1 is also associated with chylomicrons. Chylomicrons transport lipids absorbed by the intestines to the rest of the body.) Apolipoprotein B consists of two isoforms: B-48 and B-100. They're both produced from the same gene but they differ due to the editing of the resulting mRNA. B-48 received its name because it represents about 48 % of B-100. B-100 is associated with LDL cholesterol, but not with HDL cholesterol. (B-100 is also associated with VLDL particles, from which the majority of LDL is derived.) Importantly, there's only one ApoB-100 protein per lipoprotein particle. Because of this, ApoB-100 gives you insight into the number of LDL particles wandering around in the bloodstream.
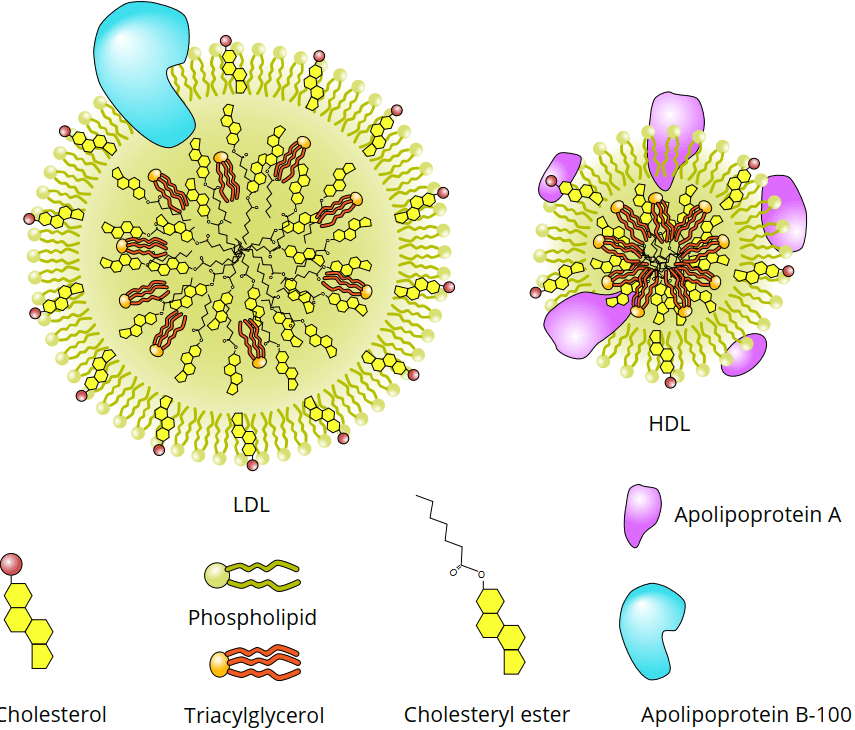
Because of this association, it might be interesting to measure their levels in the blood from a clinical perspective. In regard to apolipoprotein B, it seems to be a more potent marker of cardiovascular risk than LDL cholesterol [28]. However, the addition of this measurement to conventional risk factors (total cholesterol and HDL cholesterol) doesn't improve cardiovascular disease prediction [29]. Nevertheless, the addition of the apolipoprotein B to A1 ratio does give a slight improvement in prediction. Nevertheless, there's currently not enough evidence to know if lowering this ratio as a treatment target is effective (or more effective than conventional targeting). I suspect future research might make these measurements more popular. Either way, I've just summarized them in the table below for reference. I've calculated the ApoB to A1 ratio changes myself, as these haven't been reported in any of the trials. Therefore, no statistically significant testing has been done either, so I'm just reporting the relative change for those interested.

Lipoprotein(a) [Lp(a)]
Lipoprotein(a), or Lp(a) for short, contains the apolipoprotein ApoB-100, just like LDL cholesterol. What makes Lp(a) unique is its apolipoprotein Apo(a). And just like LDL cholesterol particles, it contributes to the process of atherosclerosis and can be viewed as a causal factor [30, 31]. While less is known about how effective treatment of Lp(a) is, with some caution, the same mantra as for LDL cholesterol can be assumed. That is, decreasing it will lower your risk of cardiovascular disease and increasing it will increase the risk.
As described in the previous sections, AAS demonstrate an unfavorable effect on serum lipids. Somewhat surprisingly, the effect on Lp(a) might be neutral to positive. That is, AAS appear to not affect or even decrease serum Lp(a). A randomized, double-blind, placebo-controlled trial evaluated the effects of nandrolone decanoate (200 mg weekly) for 8 weeks in a group of bodybuilders [15]. A 37 % decrease in Lp(a) was seen in the group receiving nandrolone, while an 18 % decrease was seen in the placebo group. Both decreases were significantly different from baseline, but not from each other. It should also be noted that there was a large range of Lp(a) values in these (small) groups. In the same paper, the authors also describe a nonblinded study with self-administered AAS and compare their results with a nonusing control group. What's interesting is that Lp(a) was greatly suppressed in these users, and after cessation, Lp(a) increased again. These results appear at odds with the randomized trial. It might be that a greater dosage of AAS leads to a larger decrease in Lp(a). Since the self-administered AAS cycles consisted of various AAS, it could also be that there's variability between the various AAS on how they affect Lp(a).
A different randomized double-blind placebo-controlled trial evaluated the effect of testosterone enanthate (600 mg weekly) for 3 weeks in elderly obese men [9]. No significant change in Lp(a) was found in the treatment group or the placebo group. If the effect on Lp(a) found in the other trial is a true effect, it might be that the 3-week period might be too short to demonstrate a measurable effect.
Either way, as it stands, AAS might decrease serum Lp(a) concentration. This might have a favorable health effect, decreasing cardiovascular disease risk. However, given the negative impact on other serum lipids, it's impossible to say that there's a net benefit with the current evidence.
References
- Caird, Francis Irvine. "Heart disease in old age." Postgraduate medical journal 39.453 (1963): 408.
- Dawber, Thomas R., Felix E. Moore, and George V. Mann. "II. Coronary heart disease in the Framingham study." International journal of epidemiology 44.6 (2015): 1767-1780.
- Ference, Brian A., et al. "Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel." European heart journal 38.32 (2017): 2459-2472.
- Jones, P. J. "Regulation of cholesterol biosynthesis by diet in humans." The American journal of clinical nutrition 66.2 (1997): 438-446.
- Bhasin, Shalender, et al. "The effects of supraphysiologic doses of testosterone on muscle size and strength in normal men." New England Journal of Medicine 335.1 (1996): 1-7.
- Bhasin, Shalender, et al. "Testosterone dose-response relationships in healthy young men." American Journal of Physiology-Endocrinology And Metabolism (2001).
- Glenney, Susan Sullivan, et al. "Effect of exercise training on cardiac biomarkers in at-risk populations: a systematic review." Journal of Physical Activity and Health 14.12 (2017): 968-989.
- Bhasin, Shalender, et al. "Older men are as responsive as young men to the anabolic effects of graded doses of testosterone on the skeletal muscle." The Journal of Clinical Endocrinology & Metabolism 90.2 (2005): 678-688.
- Herbst, Karen L., et al. "Testosterone administration to men increases hepatic lipase activity and decreases HDL and LDL size in 3 wk." American Journal of Physiology-Endocrinology And Metabolism 284.6 (2003): E1112-E1118.
- Anderson, R. A., E. M. Wallace, and F. C. W. Wu. "Effect of testosterone enanthate on serum lipoproteins in man." Contraception 52.2 (1995): 115-119.
- Thompson, Paul D., et al. "Contrasting effects of testosterone and stanozolol on serum lipoprotein levels." Jama 261.8 (1989): 1165-1168.
- Pirillo, Angela, Giuseppe Danilo Norata, and Alberico Luigi Catapano. "High-density lipoprotein subfractions-what the clinicians need to know." Cardiology 124.2 (2013): 116-125.
- Kuipers, H., et al. "Influence of anabolic steroids on body composition, blood pressure, lipid profile and liver functions in body builders." International journal of sports medicine 12.04 (1991): 413-418.
- Glazer, Gary, and Anthony L. Suchman. "Lack of demonstrated effect of nandrolone on serum lipids." Metabolism 43.2 (1994): 204-210.
- Hartgens, F., et al. "Effects of androgenic-anabolic steroids on apolipoproteins and lipoprotein (a)." British journal of sports medicine 38.3 (2004): 253-259.
- Granados, Jorge, et al. "Prohormone supplement 3β-hydroxy-5α-androst-1-en-17-one enhances resistance training gains but impairs user health." Journal of applied physiology 116.5 (2014): 560-569.
- Singh, Atam B., et al. "The effects of varying doses of T on insulin sensitivity, plasma lipids, apolipoproteins, and C-reactive protein in healthy young men." The Journal of Clinical Endocrinology & Metabolism 87.1 (2002): 136-143.
- Friedl, Karl E., et al. "High-density lipoprotein cholesterol is not decreased if an aromatizable androgen is administered." Metabolism 39.1 (1990): 69-74.
- Zmuda, Joseph M., et al. "The effect of testosterone aromatization on high-density lipoprotein cholesterol level and postheparin lipolytic activity." Metabolism 42.4 (1993): 446-450.
- Rader, Daniel J., and G. Kees Hovingh. "HDL and cardiovascular disease." The Lancet 384.9943 (2014): 618-625.
- Khera, Amit V., et al. "Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis." New England Journal of Medicine 364.2 (2011): 127-135.
- Phillips, Michael C. "Molecular mechanisms of cellular cholesterol efflux." Journal of Biological Chemistry 289.35 (2014): 24020-24029.
- Moore, Kathryn J., and Ira Tabas. "Macrophages in the pathogenesis of atherosclerosis." Cell 145.3 (2011): 341-355.
- Du, Xian-Ming, et al. "HDL particle size is a critical determinant of ABCA1-mediated macrophage cellular cholesterol export." Circulation research 116.7 (2015): 1133-1142.
- Rubinow, Katya B., et al. "Testosterone replacement in hypogonadal men alters the HDL proteome but not HDL cholesterol efflux capacity." Journal of lipid research 53.7 (2012): 1376-1383.
- Rubinow, Katya B., et al. "Sex steroids mediate discrete effects on HDL cholesterol efflux capacity and particle concentration in healthy men." Journal of clinical lipidology 12.4 (2018): 1072-1082.
- de Souza, Francis Ribeiro, et al. "Diminished cholesterol efflux mediated by HDL and coronary artery disease in young male anabolic androgenic steroid users." Atherosclerosis 283 (2019): 100-105.
- Sniderman, Allan D., et al. "A meta-analysis of low-density lipoprotein cholesterol, non-high-density lipoprotein cholesterol, and apolipoprotein B as markers of cardiovascular risk." Circulation: Cardiovascular Quality and Outcomes 4.3 (2011): 337-345.
- T. E. R. F. Collaboration*. Lipid-Related Markers and Cardiovascular Disease Prediction Lipid-Related Markers and CVD Prediction. JAMA, 307(23):2499–2506, 06
- van der Valk, Fleur M., et al. "Oxidized phospholipids on lipoprotein (a) elicit arterial wall inflammation and an inflammatory monocyte response in humans." Circulation 134.8 (2016): 611-624.
- Gencer, Baris, et al. "Lipoprotein (a): the revenant." European heart journal 38.20 (2017): 1553-1560.