In Book on Steroids there’s a passage in Chapter 2 in which I briefly describe why I think methenolone (Primobolan) is experienced as a relatively weak anabolic steroid. I ascribe this to its metabolism. Specifically, so-called 3α-reduction which is known to happen in skeletal muscle. The passage reads as follows (I’ve taken out the references):
“Since I'm talking about 3α-reduction now anyways, I'd also like to note that this is likely the reason why the AAS methenolone (Primobolan) has such relatively weak muscle-building properties. It binds about as strongly to the AR as testosterone does and in an assay that measures androgenic activity, its relative potency is even higher than that of testosterone . So how can this AAS be experienced as such a relatively weak one? Well, this assay was performed in osteosarcoma cells, not skeletal muscle cells. Skeletal muscle cells express 3α-HSD and indeed the primary metabolites of methenolone are 3α-reduced. These 3α-reduced metabolites are the result of a single chemical reaction, catalyzed by 3α-HSD. This is contrary to testosterone, which itself is a poor substrate for these enzymes. These results suggest that methenolone gets broken down to an appreciable extent in skeletal muscle, making it less potent in this tissue. So, relatively speaking, its activity in skeletal muscle (and other tissues which express 3α-HSD) gets hammered, while this doesn't occur in tissues lacking it, thus making it a bad choice for those looking for AASs with potent muscle-building effects.”
I’ve gotten some questions about it and the reason for this article in which I expand on the subject is because of a recent debate I had about it with Victor Black. In this article I try to present the available evidence with regard to this claim in a balanced way. I’ll go over what is known about methenolone’s metabolism, the pitfalls of androgen assays, and some of the clinical trials that used methenolone.
Metabolism of methenolone (Primobolan)
Before I get into the metabolism of methenolone, I briefly want to go over its structural formula. After all, its metabolism is a result of its chemical structure.
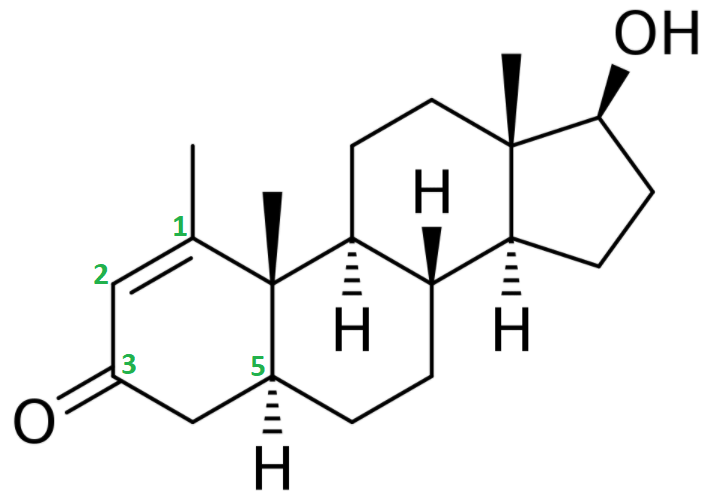
The chemical features that are of importance are the keto group (=O) at carbon 3, the α-oriented hydrogen atom (-H) at carbon 5, the methyl group (-CH3) attached to carbon 1 and the double bond (C=C) between carbon atoms 1 and 2.
Let me grind you through some organic chemistry nerd talk. Feel free to skip to the next paragraph if you don't care about it. The keto group at carbon 3 and the α-oriented hydrogen atom at carbon 5 are also seen in dihydrotestosterone (DHT). Steroids with these two chemical features generally form good substrates for enzymes that reduce the keto group at carbon 3. These enzymes are referred to as 3α/3β-hydroxysteroid dehydrogenases. The result is a hydroxyl group (-OH). The 3-keto group is very important in terms of binding affinity for the androgen receptor. Pretty much every common anabolic steroid has a 3-keto group: testosterone, nandrolone, trenbolone, boldenone, drostanolone, oxandrolone, fluoxymesterone, oxymetholone, methandienone, turinabol, etc. The reason for this is that the oxygen atom can function as a hydrogen bond acceptor for charged amino acids of the androgen receptor that lie in close vicinity of the A-ring when binding [1]. If you reduce this oxygen atom, there’s no pair of electrons to function as a hydrogen bond acceptor, and thus the binding affinity drops massively. Stanozolol might be a structural exception, since it does not have a 3-keto group (nor a hydroxyl group there), but the same thing is happening there. The second nitrogen atom of stanozolol’s pyrazole ring can function as a hydrogen bond acceptor. Anyways, enough about that. The take-home message here is that, once a steroid sees its 3-keto group reduced, it’s effectively rendered useless.
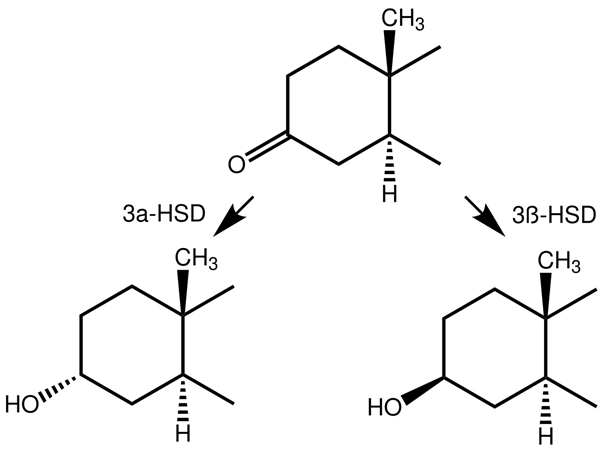
Humans express 4 different so-called isozymes that can catalyze 3α-reduction [2]. These isozymes are named AKR1C1 to AKR1C4. All are able to catalyze 3α-reduction, but differ in their tissue distribution and substrate specificity. This is in sharp contrast with the rat, that expresses only a single enzyme that’s capable of 3α-reduction. Because of the presence of multiple enzymes being able to catalyze 3α-reduction, as well as the single one in rats not being identical to any of the 4 isozymes in humans, great caution should be taken when extrapolating from rats to humans in this regard. Better yet, simply don’t.
AKR1C2 and AKR1C4 are the ones that appear to demonstrate the most 3α-reduction activity. AKR1C4 is expressed to a great extent in liver, but only to a minor extent in lung, prostate, mammary gland, brain, small intestine and testis [2]. As such, AKR1C4 seems to be a liver-specific form. AKR1C2 on the other hand is, besides being expressed in the liver, also expressed to a large extent in the lungs and prostate. Some smaller, but significant, expression occurs in other tissues as well.
Of course, I hear you thinking: “What about skeletal muscle?” Good question. Unfortunately, the study I mentioned above did not check for this. Another study did check for presence in skeletal muscle, but only did so for AKR1C1 and AKR1C4 [3]. Both could not be detected in skeletal muscle. Yet another study found AKR1C3 to be expressed in muscle, and found that its mRNA expression is upregulated with testosterone therapy (250 mg Sustanon biweekly) [4]. I have failed to find any study examining the expression of AKR1C2 in skeletal muscle.
So far we have established that at least one, and possibly two, of the isozymes that can 3α-reduce steroids are expressed in skeletal muscle. Is there then also evidence that demonstrates significant activity of these enzymes in skeletal muscle? An old German paper elegantly demonstrated this by intravenously injecting labeled DHT to three participants in order to track its metabolic fate [5]. Muscle tissue samples were collected 20 to 60 minutes after injection. 29.4-51.0 % of the labeled steroid was identified as 3α-androstanediol (the 3α-reduced metabolite of DHT) in these samples. A couple of other metabolites were also identified and only 18.1 to 26.2 % of DHT was left. This indeed demonstrates significant breakdown of DHT occuring in skeletal muscle tissue as a result of reduction of its 3-keto group. As a side note, they did the same thing with labeled testosterone, of which 69.5-79.8 % of the labeled steroid was still identified as testosterone in the muscle tissue samples.
Now what about methenolone (Primobolan)? As mentioned at the start of this section, it also has a keto group at carbon 3 and an α-oriented hydrogen atom at carbon 5. The reason why it does not necessarily follow the same fate as DHT, is because of the other 2 alterations: the methyl group attached to carbon 1, and the double bond between carbon 1 and 2.
What is known about methenolones metabolism? The following table was brought to my attention by Victor Black. It stems from a study done by the manufacturer of methenolone (Schering, nowadays Bayer) [6].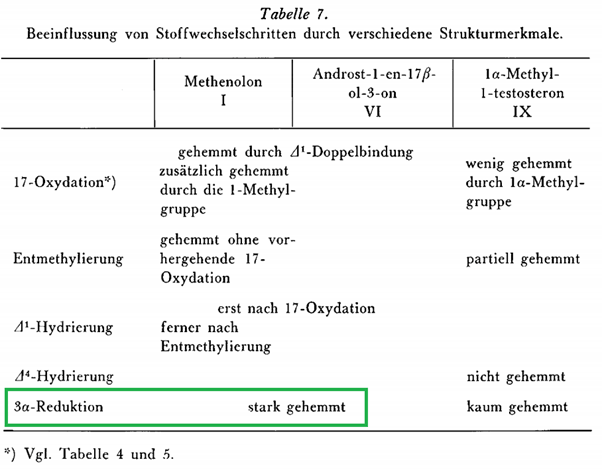
The words ‘stark gehemmt’ are German for ‘strongly inhibited’. As such, this table suggests the 3α-reduction of methenolone is strongly inhibited. This would follow from table 5 from the same paper. Table 5 lists 2 potential 3α-reduced metabolites of methenolone that they checked for. Indeed, one of them, 1-(α)-Methyl-androstan-3α-ol-17-on (methylandrosterone), was listed as undetectable. The other, androstan-3α-ol-17-on (androsterone), was listed as (+), which I suppose means they found a trace amount. Methenolone itself was retrieved to the largest extent, listed as +++ in the table. (Undoubtedly this was conjugated, as later research was unable to retrieve unchanged methenolone from urine after injection [8].)
So what are these two metabolites? Methylandrosterone is a 3α-reduced form of methenolone that lacks the double bond between carbon 1 and 2. The other one, androsterone, additionally lacks the 1a-methyl group.
But these aren’t the only potential 3α-reduced metabolites of methenolone. Indeed, later research demonstrated that after oral administration, the primary metabolite of methenolone was 3α-hydroxy-1-methylen-5a-andorstan-17-one [7]. In fact, it got excreted to roughly a 50 % larger extent than (conjugated) methenolone itself. As such, the note that 3α-reduction is ‘strongly inhibited’ in the table above is based on incomplete screening of metabolites and should be discarded as false given the research that came out later.
However, they note that they extracted 1.62-1.65% of the ingested methenolone as unchanged in the urine and 2.4% of the excreted methenolone dose as the 3α-reduced metabolite (3α-hydroxy-1-methylen-5α-andorstan-17-one) within 7 days after ingestion. They don't mention the % of other metabolites, but it can be derived they cumulatively made up no more than 1% of the methenolone dose. All in all, about 95 % of the ingested dose remains unaccounted for. This can be explained by two factors:
- Missing additional metabolites
- Excretion through other routes (faeces through entrohepatic recycling)
Future research using a state-of-the-art technique (LiQuiD ChRoMaToGrApHy QuAdRuPoLe TiMe-Of-FliGhT mAsS sPeCtRoMeTry aka LC–QTOFMS) indeed found several additional metabolites after methenolone injection [8]. However, they did not perform a quantitative analysis, so it cannot be derived for how much of the missing % of the methenolone dose this accounts. It would be my guess that the majority ends up in the faeces rather than that undetected metabolites comprise the majority of the unaccounted dose.
Also, study [8] did not find literally unchanged methenolone back in the urine (they did find conjugated methenolone without other chemical alterations). This means the unchanged methenolone from other studies needs to have been a mix-up with a different metabolite, or the sulfo or glucuronic acid groups were removed post-collection of the urine sample. (It's most likely the latter, since they used GC-MS.)
Either way, the take-home message is that methenolone does get significantly 3α-reduced, but to a lesser degree than DHT. The unanswered questions are to what extent this happens in skeletal muscle (given the different isozymes) and at what rate. These questions are unanswerable with the current data.
Androgen assays
Given that methenolone gets 3α-reduced, and skeletal muscle expresses enzymes that can catalyze this reaction, it can be argued that methenolones effect in skeletal muscle gets diminished. It is uncertain to what degree this happens. Additionally, other androgen-sensitive tissues that express enzymes that can do this will also diminish its effect in these tissues. It can therefore be argued that in some androgen-sensitive tissues its effect will be diminished too. What I’m trying to say here is that on the one hand it can be detrimental for its so-called anabolic to androgenic ratio, whereas on the other hand it might be beneficial — depending on the tissue.
Quantifying the anabolic to androgenic ratio for AAS is something certain assays have attempted to do. The primary example of such an assay is the Hershberger assay. This assay was first described by Hershberger and his colleagues in 1953 [9]. You take some rats and castrate them to get rid of any endogenous testosterone produced that might interfere with your results. Then you inject them with the AAS under investigation and you wait a while (originally 8 days) before you kill the rats and dissect them. You dissect them in order to weigh the levator ani muscle, the ventral prostate and the seminal vesicles. The weight increase of the levator ani muscle is proposed to be reflective of the compound’s anabolic activity and the weight increase of the ventral prostate and seminal vesicles is supposed to be reflective of its androgenic activity.
The problems with this assay are numerous. First, picking the levator ani muscle as a proxy for skeletal muscle is faulty and has received quite some criticism from the scientific community. This muscle, as it turns out, is not an ordinary skeletal muscle. It’s an androgen-dependent muscle that is part of the reproductive system of the rat (it’s actually the dorsal bulbocavernous muscle). A paper by Keith Hayes extensively covers this [10]. In a sense, you could thus argue that a weight increase of the dorsal bulbocavernous muscle is representative of androgenic activity rather than anabolic activity.
Another problem is that the dorsal bulbocavernous muscle and seminal vesicles respond differently to varying concentrations of an androgen. As such, the measured ratio will be different depending on the dose used as well as the point in time measurements were taken. This is illustrated well in the following figure taken from van der Vies after injection of nandrolone [11].
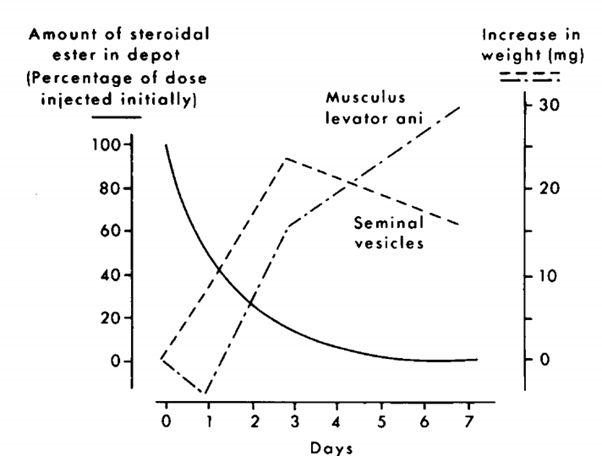
During the first 3 days, the concentration is high enough to stimulate growth of both the seminal vesicles as well as the dorsal bulbocavernous muscle. However, after three days the concentration is not high enough to maintain this growth for the seminal vesicles, and they decrease in size again. Nevertheless, the dorsal bulbocavernous muscle is still stimulated sufficiently to continue growing in size. As such, the anabolic to androgenic ratio that you’d measure on day 3 would be a whole lot different from what you would measure at day 7, despite it being the same compound. This also highlights that different organs simply respond differently depending on the concentration of the compound. Even if there would be an accurate way of determining an anabolic to androgenic ratio, extrapolating it from physiological concentrations to supraphysiological concentrations would be completely flawed!
Continuing with this, it is a flawed assumption that growth of the ventral prostate or seminal vesicles is representative of all other androgenic effects. There is no evidence to support this assumption (on the contrary).
Finally, rats aren’t humans. It’s another huge assumption that the homologous tissues in humans would respond in the same way to a steroid as in the rat. No value should be attributed to the ratios coming from the Hershberger assay.
Another method is through measuring the relative binding affinity of a steroid in various tissues. Saartok et al. used this method by determining the relative binding affinity of methenolone in rabbit muscle, rat muscle and rat prostate, compared to the reference compound (methyltrienolone) [12]. The authors were aware of the problems with the dorsal bulbocavernous muscle and appear to have used the soleus and gastrocnemius muscles instead. The results were as followed:
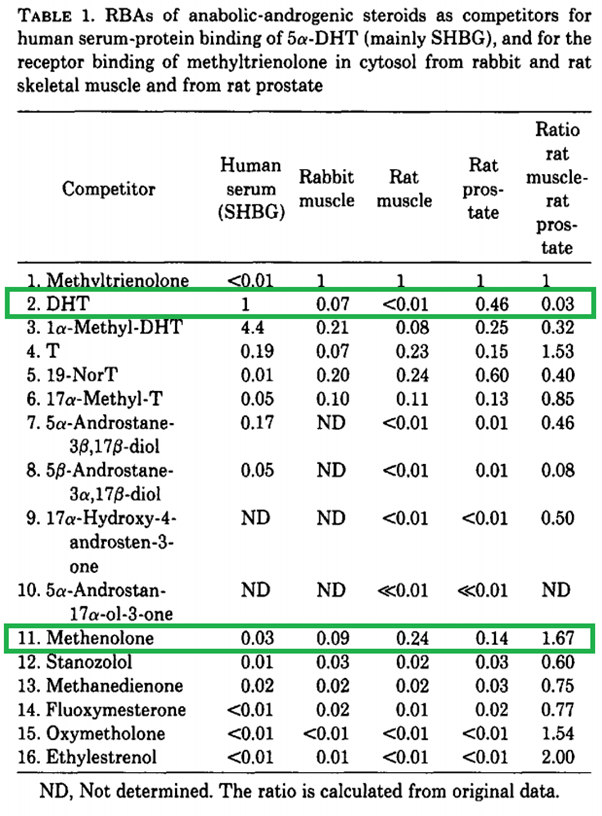
Let’s first take a look at DHT and compare it with T (testosterone). In rat muscle, its binding affinity is way lower than that of testosterone. This is most likely because of rapid metabolization through 3α-reduction in skeletal muscle (as also speculated by the authors). However, the binding affinity is similar to that of testosterone in rabbit muscle. This highlights a notable difference between two animal species. Apparently significantly less 3α-reduction of DHT takes place in rabbit skeletal muscle. Now, if you look at methenolone, the binding affinities are in the same ballpark as those of testosterone. Does this mean it does not get 3α-reduced in humans? Absolutely not. As mentioned earlier in this article, there are different enzymes at work which differ in their substrate specificity compared to rats, and this study alone already shows a notable difference between two animal species when you look at the numbers of DHT.
The uselessness of this assay for getting an idea of the anabolic to androgenic ratio (or the anabolic activity alone) in humans is further highlighted by the ridiculously low binding affinities of some of the other AAS.
A final assay I would like to discuss is the androgen receptor (AR) CALUX assay [13]. The assay was developed to detect novel doping substances in athletes rather than be used to derive these ratios or a compound’s anabolic activity from. For the purpose of detecting novel doping substances, it’s perfect. Basically they’ve modified a cell in such a way that something lights up when the androgen receptor is activated. They’ve done this by co-transfecting the cell with a luciferase gene that’s under transcriptional control of the androgen receptor.
For this purpose, they’ve used osteosarcoma cells. I’m gonna keep this brief, because a main issue with this with regard to methenolone is that these cells clearly do not have any significant 3α-reduction activity. This is evidenced by the fact that DHT pops up as a massively potent AAS in this assay. So you can look up what they measured with methenolone, but it’s not gonna tell you anything about what’s actually happening in skeletal muscle.
Clinical trials
In the end, what you’d want to know is how methenolone stacks up to another steroid in a clinical trial in terms of side effects and muscle growth. Do head-to-head trials exist? Yeah, but done in breast cancer patients without measuring muscle growth in an awkwardly irrelevant clinical setting for bodybuilders. Another thing you can do is look at what they find in clinical trials without a direct comparison and lay this next to other trials which used other compounds. Admittedly, this is already a failed endeavour as comparisons between trials — especially with smaller sample sizes — is very troublesome.
There’s one trial that could’ve been awesome. 47 men were randomized to receiving a placebo, methenolone acetate, placebo + training, and methenolone acetate + training [14]. The study lasted a whopping 16 weeks. They performed strength measurements and weighed the subjects. So why wasn’t it awesome? Because they gave the methenolone (20 mg daily) orally instead as injections. While it does have some oral bioavailability, it’s definitely not up par with that of the 17a-alkylated AAS. As such, it gets metabolized too quick to exert an effect at that dosage. Indeed, the authors found no differences between groups in any of the measurements.
Another trial examined the effect of 100 mg methenolone enanthate by intramuscular injection once a week for 6 weeks [15]. That sounds good. However, it was done alongside rehabilitation therapy in patients with hemiplegia 1-8 months after stroke. That is, patients with complete paralysis of half the body. (Obviously the rehabilitation therapy aimed at the nonparalysed half of the body.) The study was nonblinded and nonrandomized and the participants in the placebo and methenolone groups were old folks (on average 62 to 67 years of age). Strength measurements were up and so was muscle cross sectional area of the thighs. It’s hard - if not impossible - to say how it performs compared to other AAS. The culprit with methenolone is not so much that it doesn’t work: it does. The culprit is that its activity in skeletal muscle might get diminished, making it suboptimal to put it gently.
There’s a trial with (female) breast cancer patients receiving methenolone enanthate by intramuscular injection [16]. The women first received 3x 400 mg weekly, after which the dosage got reduced to 3x 200 or 3x 300 mg weekly. Now that’s a dosage. The therapy lasted, on average, 7.7 months in these women. There is little that can be derived from this paper, other than that methenolone increases total cholesterol quite significantly. This is something common with orally bioavailable AAS. And perhaps this effect might be more pronounced in women, I don’t know.
Anyways, I found one trial in which they did a head-to-head comparison in patients with advanced breast cancer [17]. One group received a very high dosage of methenolone (3x 400 mg weekly) and the other received a high dosage of testosterone propionate (3x 100 mg weekly). 48 % of the patients in the methenolone group had objective improvement of their breast cancer (and clearly a longer time of survival), whereas no one showed improvement in the testosterone group.
This is all nice and all, but this doesn’t bring us further with regard to how it compares to other compounds in terms of muscle-building.
If someone is aware of some obscure old clinical head-to-head trial of methenolone compared to another AAS in healthy folks, in which body composition or strength was measured, please contact me.
Conclusions
So far we've established a few things.
Methenolone is broken down to an appreciable extent by means of 3α-reduction, as evidenced by its primary metabolite 3α-hydroxy-1-methylen-5a-andorstan-17-one. Once reduced, it doesn’t do much anymore. There are 4 isozymes in humans that are distinct from the single enzyme in rats that catalyzes this reaction and vary from each other in terms of substrate specificity and tissue distribution. This makes extrapolating from rats (or any other animal for that matter) flawed. Of these 4 isozymes, at least 1 is expressed in skeletal muscle, and possibly 2. While methenolone does get 3α-reduced, it does so to a lesser extent than DHT. It is unknown to what degree this happens in skeletal muscle and at what rate. Current data is insufficient to answer these important questions.
Androgen assays are nice, but they all have significant flaws, making them notoriously unreliable to rely on to say whether a certain compound is more anabolic or androgenic than another in practice. As such, reliable information with regard to methenolone cannot be derived from these studies.
There are a few clinical trials done with methenolone, and one in particular would've been wildly interesting &emdash; if they didn't give the methenolone orally instead of injecting it. Lack of head-to-head trials with healthy participants in which body composition or muscle strength is measured makes answering any questions about how methenolone relates to other AAS impossible.
To throw in my opinion: most people have described methenolone to me as a relatively mild AAS. Implying that they need to stack it with other AAS, or dose it high, for an appreciable effect on building muscle mass. This was the main reason for me to find a cause for this apparent effect: people told me it was a mild AAS, and I found 3α-reduction in skeletal muscle the most likely explanation. However, I’m sure other people disagree with this description of methenolone’s effect and/or what I make from that.
Finally, whether you like it or not, evidence is mounting that whatever AAS is on the label of your vial, is most likely not in it. (Unless you got it from a pharmacy, but this isn’t particularly likely with methenolone to begin with.) A recent study conducted by the outpatient clinic for anabolic steroid users in the Netherlands examined 272 AAS samples from 46 different brands supplied to them by the subjects who enrolled in their clinical trial [18]. The results can be found in the table taken from the paper below:
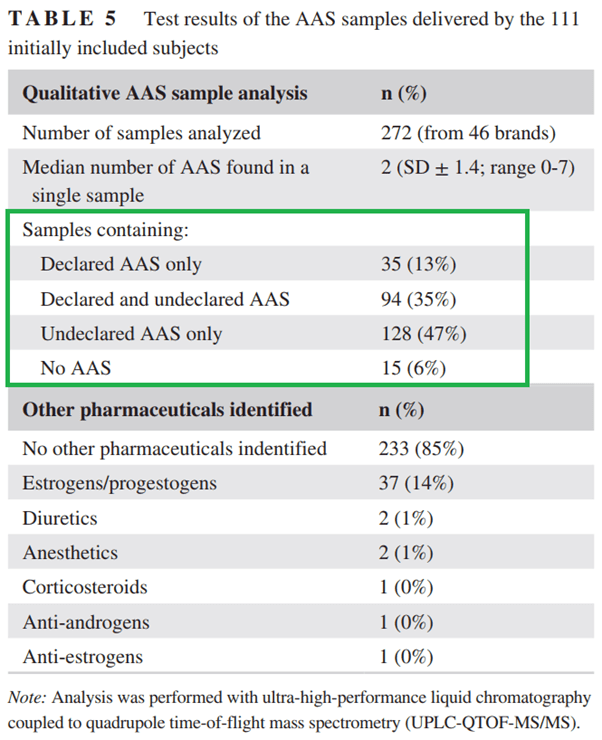
Shockingly, only 13% of the supplied AAS only had the declared AAS in them. And mind you, this was a qualitative analysis. So this doesn’t even mean these were dosed as declared on the label!
And I hear you thinking: “Yeah but those are losers, mine is real, I got a trustworthy supplier.” Do you? I know some of the subjects which participated in this trial personally. They all were super convinced that their AAS were good to go as described on the label. It was unthinkable to them that their stuff wasn’t legit. They had the best supplier in town. Nevertheless, they were wrong.
The authors of the paper also mention this: “Attempting to circumvent untrustworthy AAS products by buying from a seemingly “reliable” brand or dealer—an everintriguing topic to users of AAS—appears trifling as no brand showed consistent reliability.”
Similar results were published in 2005 by the Dutch doping authority after examination of 336 products [19] and in Sweden when they analysed over a 1.000 samples of doping products seized at their border [20].
The shadiness of the AAS black market adds to the unreliability of personal accounts of AAS usage beyond the obvious drawbacks of anecdotes. Most AAS users simply aren’t taking whatever they think they’re taking.
References
- Pereira de Jésus-Tran, Karine, et al. "Comparison of crystal structures of human androgen receptor ligand‐binding domain complexed with various agonists reveals molecular determinants responsible for binding affinity." Protein Science 15.5 (2006): 987-999.
- Penning, Trevor M., et al. "Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1–AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones." Biochemical journal 351.1 (2000): 67-77.
- O'CONNOR, Tania, et al. "Major differences exist in the function and tissue-specific expression of human aflatoxin B1 aldehyde reductase and the principal human aldo-keto reductase AKR1 family members." Biochemical journal 343.2 (1999): 487-504.
- Gharahdaghi, Nima, et al. "Testosterone therapy induces molecular programming augmenting physiological adaptations to resistance exercise in older men." Journal of Cachexia, Sarcopenia and Muscle 10.6 (2019): 1276-1294.
- Becker, H., et al. "In vivo uptake and metabolism of 3H-testosterone and 3H-5α-dihydrotestosterone by human benign prostatic hypertrophy." European Journal of Endocrinology 71.3 (1972): 589-599.
- Langecker, H. "BEZIEHUNGEN ZWISCHEN SUBSTITUTION IM RING A UND ABBAU IM STOFFWECHSEL BEI VERWANDTEN DES TESTOSTERONS." European Journal of Endocrinology 41.4 (1962): 494-506.
- Goudreault, Danielle, and Robert Massé. "Studies on anabolic steroids—4. Identification of new urinary metabolites of methenolone acetate (primobolan®) in human by gas chromatography/mass spectrometry." The Journal of steroid biochemistry and molecular biology 37.1 (1990): 137-154.
- He, Genye, et al. "New long term metabolite in human urine for metenolone misuse by liquid chromatography quadrupole time-of-flight mass spectrometry." Steroids 105 (2016): 1-11.
- Hershberger, L. G., Elva G. Shipley, and Roland K. Meyer. "Myotrophic activity of 19-nortestosterone and other steroids determined by modified levator ani muscle method." Proceedings of the Society for Experimental Biology and Medicine 83.1 (1953): 175-180.
- Hayes, Keith J. "The so-called 'levator ani' of the rat." European Journal of Endocrinology 48.3 (1965): 337-347.
- Van der Vies, J. "Implications of basic pharmacology in the therapy with esters of nandrolone." European Journal of Endocrinology 110.3_Suppla (1985): S38-S44.
- Saartok, Tönu, Erik Dahlberg, and Jan-Åke Gustafsson. "Relative binding affinity of anabolic-androgenic steroids: comparison of the binding to the androgen receptors in skeletal muscle and in prostate, as well as to sex hormone-binding globulin." Endocrinology 114.6 (1984): 2100-2106.
- Houtman, Corine J., et al. "Detection of anabolic androgenic steroid abuse in doping control using mammalian reporter gene bioassays." Analytica chimica acta 637.1-2 (2009): 247-258.
- Fowler JR, William M., Gerald W. Gardner, and Glen H. Egstrom. "Effect of an anabolic steroid on physical performance of young men." Journal of Applied Physiology 20.5 (1965): 1038-1040.
- Shimodozono, Megumi, et al. "Addition of an anabolic steroid to strength training promotes muscle strength in the nonparetic lower limb of poststroke hemiplegia patients." International Journal of Neuroscience 120.9 (2010): 617-624.
- Garbrecht, M., et al. "Hyperlipoproteinämie bei additiver Behandlung des metastasierenden Mammakarzinoms mit Metenolonönanthat." DMW-Deutsche Medizinische Wochenschrift 106.13 (1981): 400-403.
- Kennedy, B. J., and John W. Yarbro. "Effect of methenolone enanthate (NSC‐64967) in advanced cancer of the breast." Cancer 21.2 (1968): 197-201.
- Smit, Diederik L., et al. "Baseline characteristics of the HAARLEM study: 100 male amateur athletes using anabolic androgenic steroids." Scandinavian Journal of Medicine & Science in Sports 30.3 (2020): 531-539.
- https://www.dopingautoriteit.nl/media/files/documenten/Kwaliteit%20Illegale%20Dopingmiddelen.pdf (Retrieved 17-09-2020)
- Weber, Christina, et al. "Qualitative and semiquantitative analysis of doping products seized at the Swiss border." Substance use & misuse 52.6 (2017): 742-753.